Skyline 25.2 will have a new feature "Peak Imputation" which will improve several scenarios where Skyline has traditionally chosen incorrect peaks.
As of June 26, 2025, this feature is not yet part of Skyline-daily, but can be used in a special build:
https://proteome.gs.washington.edu/~nicksh/SpecialSkylines/PeakImputation/
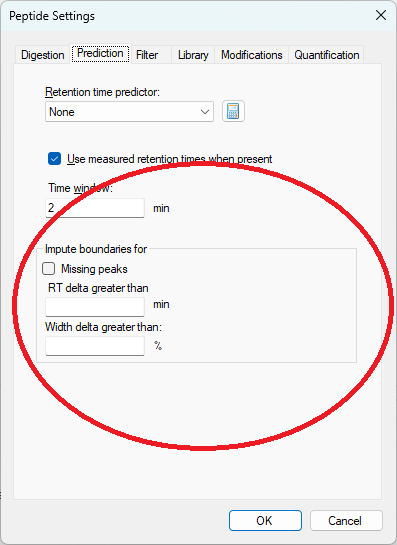
The new peak imputation settings can be found on the "Prediction" tab in Peptide Settings
The peak imputation settings tell Skyline that in certain circumstances, the peak boundaries should be chosen instead based on the peak boundaries in the best-scoring replicate for that peptide or molecule.
Skyline has two different ways of choosing peak boundaries:
When Skyline is doing its own peak detection and scoring, the peak imputation feature must be invoked as a separate step after all results have been imported.
After the peak imputation settings have been specified on the "Prediction" tab in Peptide Settings, the menu item "Refine > Reintegrate" must be used to tell Skyline to re-pick peaks using Skyline's peak scoring model combined with the peak imputation settings
When Skyline is performing this reintegration, if Skyline sees that the peak boundaries in a particular Replicate are more than the "RT delta greater than" setting away from the best-scoring replicate, or the peak width is more different than the "Width delta greater than" setting, then Skyline will move the peak boundaries of the lower-scoring replicate so that they exactly match the best-scoring replicate.
Some types of libraries, including DIA-NN and EncyclopeDIA, specify start and end times of peak integration boundaries which Skyline uses instead of Skyline's own peak detection and scoring algorithms.
When Skyline is using the peak boundaries from the library, Skyline will apply the peak imputation settings when chromatograms are first extracted. It is not necessary to do the separate step of "Refine > Reintegrate" (also, "Refine > Reintegrate" does not do anything in a document which used peak boundaries from the library because there is only one candidate peak to choose from in any particular replicate).
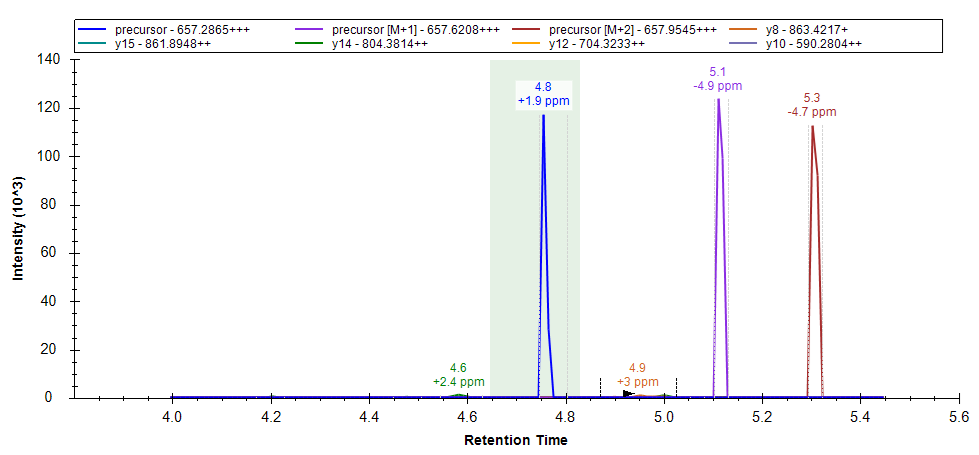
The chromatogram graph displays as a green-shaded rectangle, the peak boundaries from the best-scoring replicate
Instead of using the exact peak boundaries from the best-scoring replicate, Skyline performs a retention time alignment to map the times from the best-scoring replicate to the target replicate. If there is an iRT calculator in the document, then the iRT calculator is used to do the alignment. If there is no iRT calculator, but there is exactly one spectral library in the document, then runs are aligned using a lowess regression against the median retention time values from that spectral library.
| Attached Files | ||
PeakImputationSettings.png ImputedBounds.png| previousnext |
| expand allcollapse all |